Investigating microbial species composition and activity in the ruminant animal under different dietary conditions to determine how dietary intervention can be used to reduce methane emission in ruminants
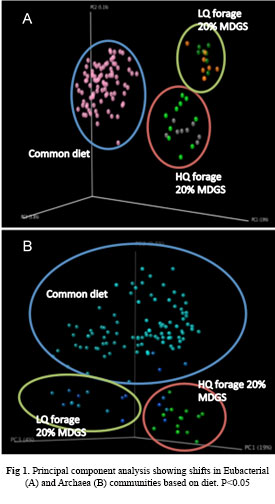 Among the major sources of methane production, ruminants account for a considerable fraction of the anthropogenic methane produced, where, enteric fermentation by ruminants is considered the single largest source of methane production worldwide. At the heart of anaerobic methane production in ruminants is a microbial food chain. The microscale processes of this microbial food chain are greatly influenced by diet. However, the interactions between diet, microbial community composition, and methane emission are poorly understood. Consequently, I am interested in understanding the interactions between diet, gut microbiota composition and methane emission, to develop science-based dietary intervention strategies to reduce greenhouse gas emission from cattle in intensive and extensive production systems. To this end, our preliminary data show that microbial community shifts in methanogenic populations can be attained by diet (Fig 1). To further understand the rumen methanogenic population and to manipulate the microbial community to reduce methane emission, I will use multiple culture independent DNA based molecular approaches to understand rumen microbial ecology and structure function relationships that include phenotyping the gut microbiota using 16S rRNA gene sequencing, metagenome sequencing, and meta-transcriptome sequencing, stable isotope probing, single cell genomics and bioinformatic approaches.
Among the major sources of methane production, ruminants account for a considerable fraction of the anthropogenic methane produced, where, enteric fermentation by ruminants is considered the single largest source of methane production worldwide. At the heart of anaerobic methane production in ruminants is a microbial food chain. The microscale processes of this microbial food chain are greatly influenced by diet. However, the interactions between diet, microbial community composition, and methane emission are poorly understood. Consequently, I am interested in understanding the interactions between diet, gut microbiota composition and methane emission, to develop science-based dietary intervention strategies to reduce greenhouse gas emission from cattle in intensive and extensive production systems. To this end, our preliminary data show that microbial community shifts in methanogenic populations can be attained by diet (Fig 1). To further understand the rumen methanogenic population and to manipulate the microbial community to reduce methane emission, I will use multiple culture independent DNA based molecular approaches to understand rumen microbial ecology and structure function relationships that include phenotyping the gut microbiota using 16S rRNA gene sequencing, metagenome sequencing, and meta-transcriptome sequencing, stable isotope probing, single cell genomics and bioinformatic approaches.
Uncovering and understanding the ecological role of viruses within the ruminant ecosystem
Viruses are the most numerous biological entities on the planet, yet little is known about their impact on community structuring due to their size, rapid evolution, and the fact that the majority cannot be cultured. The role of viruses in structuring microbial communities has been described using the kill the winner theory, constant diversity dynamics, and periodic selection using simulations. Yet, how viruses structure bacterial communities in the gut and what role they play in ecosystem efficiency is poorly understood. With contemporary metagenomic approaches the study of viral properties is now a possibility, which has helped in determining the influence of viral populations on complex environments such as the gut microbiome. We are using molecular approaches to better influence rumen bacterial communities, and ecosystem function and are testing ecological theories within the gut to predict ecosystem efficiency. As a first step towards identifying the rumen virome, we have isolated and sequenced the rumen virome under different dietary conditions to identify how viruses help adapt bacterial communities to changing diets (fig 2). Further studies in understanding the viral community within the rumen will lead to the development of new ecological theory to explain the role of viruses in structuring the microbial community and the role of viruses in the rumen.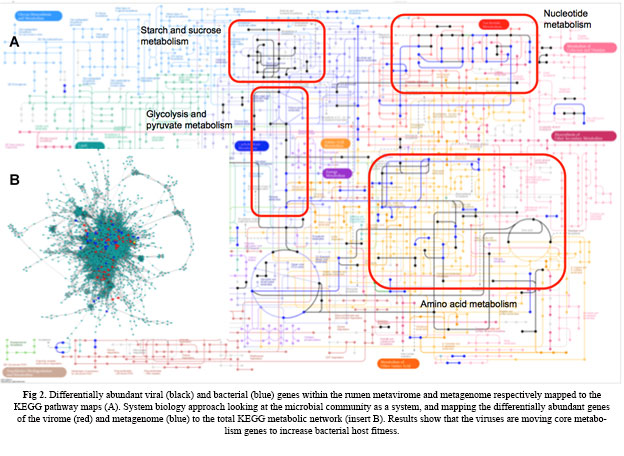
Develop a cfecum cannulated human gut microbiota associated pig model by inter-species transplantation of gut microbiota from humans to pigs to understand the effect of microbial activity on host function
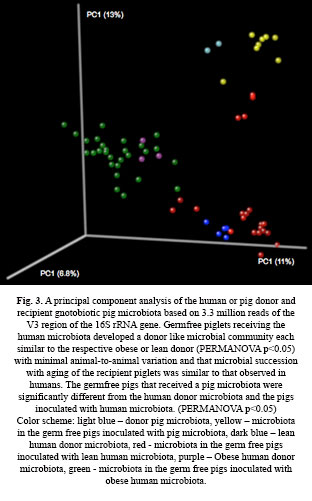 Recent studies have demonstrated that the human microbiome to play an important role in human health and it has been implicated to conditions such as obesity, inflammatory bowel disease, diabetes, metabolic syndrome, and autism. The population structural shifts of the gut microbial community in obesity and other diseases are well characterized. Although these studies have helped us understand changes in microbial community structure, and the metabolic, and the metabiome that ameliorate the disease phenotype. Partly, this is due to the lack of a good model system that allows meta-transcriptomic analysis and a metabolomic analysis of the human microbiome over time at the site of action. We have established a movel humanized pig model by inter-species transplantation of gut microbiota from humans to pigs and demonstrate that a human microbiota can be maintained with the pig (fig 3). This model has the potential to understand the effect of microbial activity on host function. Specifically, microbial regulation of host genes over time without the complications of the current mouse models that fail to develop the full repertoire of immune responses and colonization of certain Bifidobacterial species. This model will provide new opportunities to study the human microbiome during pathogen invasion (Clostridium difficile infection), obesity, metabolic disease and many other metabolic diseases.
Recent studies have demonstrated that the human microbiome to play an important role in human health and it has been implicated to conditions such as obesity, inflammatory bowel disease, diabetes, metabolic syndrome, and autism. The population structural shifts of the gut microbial community in obesity and other diseases are well characterized. Although these studies have helped us understand changes in microbial community structure, and the metabolic, and the metabiome that ameliorate the disease phenotype. Partly, this is due to the lack of a good model system that allows meta-transcriptomic analysis and a metabolomic analysis of the human microbiome over time at the site of action. We have established a movel humanized pig model by inter-species transplantation of gut microbiota from humans to pigs and demonstrate that a human microbiota can be maintained with the pig (fig 3). This model has the potential to understand the effect of microbial activity on host function. Specifically, microbial regulation of host genes over time without the complications of the current mouse models that fail to develop the full repertoire of immune responses and colonization of certain Bifidobacterial species. This model will provide new opportunities to study the human microbiome during pathogen invasion (Clostridium difficile infection), obesity, metabolic disease and many other metabolic diseases.